

FPGA-Accelerated Smith-Waterman Systolic Array for DNA Sequence Alignment
Abstract
Abstract
Aim
To design and implement a reconfigurable hardware architecture that accelerates the Smith-Waterman local alignment algorithm, overcoming the computational bottlenecks of software-based bioinformatics tools.
Introduction
Sequence alignment is a fundamental task in bioinformatics used to identify functional and evolutionary similarities between DNA or protein sequences. While the Smith-Waterman (SW) algorithm provides optimal local alignment, its O(n²) quadratic complexity makes it computationally expensive for large genomic databases. This project implements a systolic array architecture on an FPGA to perform fine-grain parallel processing, significantly reducing execution time compared to general-purpose processors.
Literature Survey and Technologies Used
Literature Survey: The implementation is based on the modified SW equations proposed by Jiang et al., which improve mapping efficiency to FPGA fabric by introducing interim variables to balance the critical path.
Hardware: Xilinx Basys 3 FPGA (Artix-7), Block RAM (BRAM) for sequence storage.
Software: Verilog HDL for RTL design, Vivado for Synthesis and Implementation.
Algorithm Parameters: Supports customized scores (Match: +2, Mismatch: -1, Gap: -1) and a 20-bit word width for the maximum similarity score.
Methodology
The system is built using a hierarchical modular approach:
Processing Element (PE): The core unit that computes the similarity score H(i,j) based on the scores from its left, upper, and diagonal neighbors. It uses a 20-bit signed format to handle peak values.
Systolic Array (Genvar_wrapper): A linear array of PEs where the query sequence is loaded into the array, and the subject sequence is streamed through it.
Max Score Logic: A dedicated module that monitors all scores in the array at every clock cycle to track the peak alignment score and its corresponding indices (best(i), best(j)).
Control FSM (Top.v): Manages the multi-phase operation: resetting the array, loading the query from BRAM, and streaming the subject sequence while tracking the global maximum.
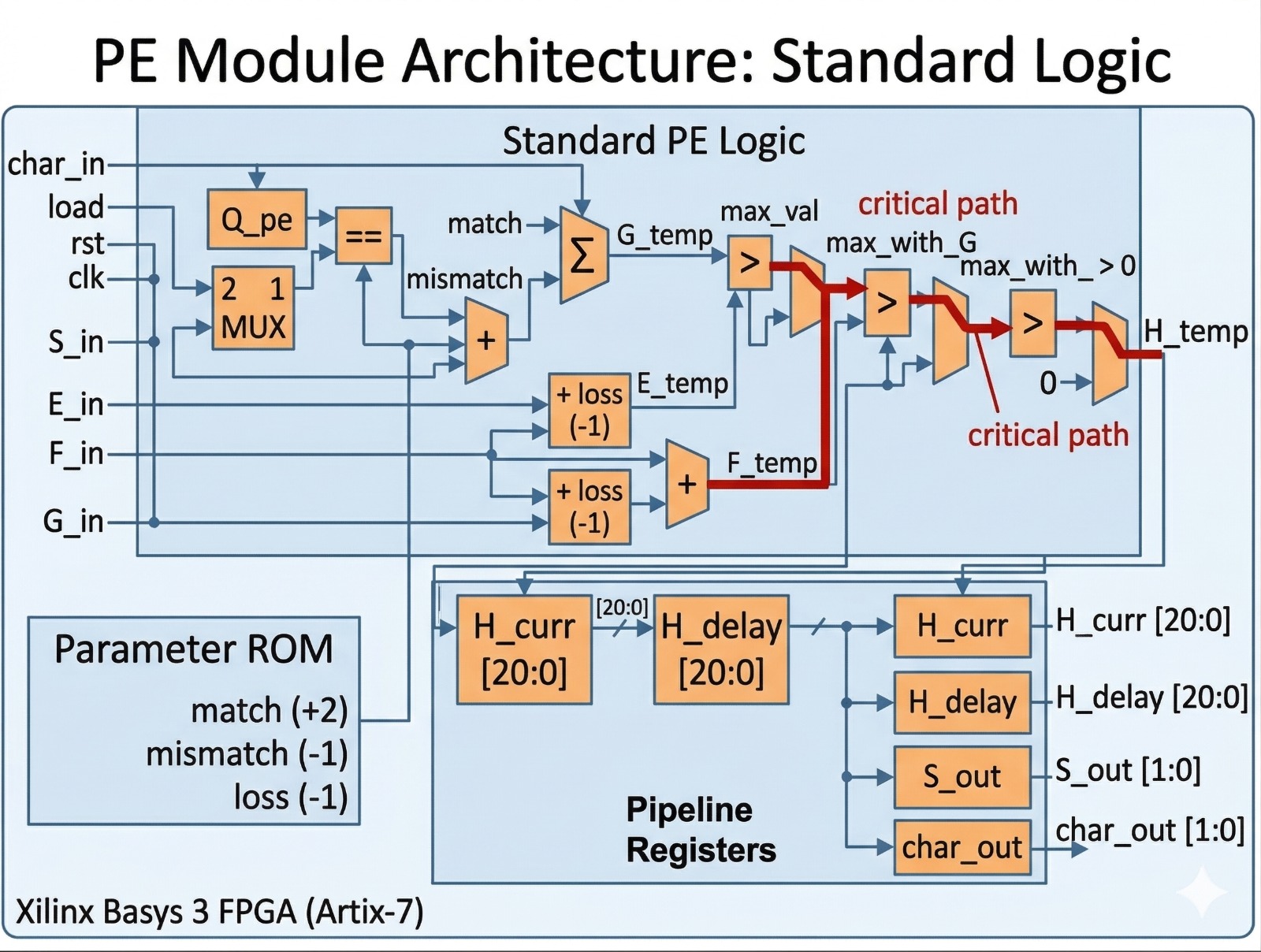
Methodology: Systolic Array Architecture
The core of this implementation is a 1D Systolic Array, which transforms the traditional O(n²) space-intensive Smith-Waterman matrix into a hardware-efficient linear pipeline.
Spatial Optimization: Instead of storing a full m*n matrix in memory, this design utilizes a linear array of m Processing Elements (PEs), where m is the length of the query sequence.
Systolic Pipelining: The subject sequence is streamed through the array character-by-character. Each PE calculates one cell of the matrix per clock cycle and passes the results to its neighbor, allowing for massive parallel execution of diagonal matrix cells.
Path Balancing: Following the Jiang et al. paper, the PE logic is modified to balance the critical path by introducing interim variables (H_delay), which reduces the delay from three "max" operations to two, enabling higher clock frequencies.
Hardware Rationale: Software implementations on general-purpose CPUs are bottlenecked by sequential processing. This FPGA implementation provides a significant speedup by performing simultaneous "cell updates" across the entire PE array.
Step-by-Step Module Workflow
The following table explains how the specific modules in your code coordinate to perform the alignment:
|
Step |
Module | Functional Action |
|
1. Reset & Global Sync |
Top.v | The FSM initializes the system, setting the awake and process flags to zero and clearing the address registers for BRAM. |
| 2. Query Retrieval | Top.v | The system reads the query sequence from ram_seq1 and flattens it into a single bit-vector (flattened_query) to be sent to the PE array. |
| 3. Parallel Loading | Genvar_wrapper.v | When the load signal is pulsed, every PE in the array simultaneously captures its designated query character into its internal Q_pe register. |
| 4. Subject Streaming | Top.v | The subject sequence is fetched from ram_seq2 and streamed into the first PE (PE_GEN[0]) at a rate of one character per clock cycle. |
| 5. Distributed Calculation | PE.v | Each PE computes the local alignment score by comparing its Q_pe with the incoming S_in. It factors in the scores from its left neighbor (E_in) and diagonal neighbor (G_in). |
| 6. Zero-Floor Logic | PE.v | The PE applies the Smith-Waterman "zero floor" (max_with_G > 0), ensuring that negative similarity scores are reset to zero to support local alignment. |
| 7. Systolic Transfer | PE.v | n every clock edge, the PE shifts the subject character (S_out) and its current score (H_curr) to the next PE in the chain. |
| 8. Global Max Tracking | Max.v | This module monitors the all_scores bus, which aggregates the H_curr value from every PE. It performs a parallel comparison to find the highest score and its PE index (best_i). |
| 9. Result Reporting | Top.v | Once the subject sequence has fully traversed the pipeline, the FSM asserts done and outputs the final peak score and its coordinates in the alignment. |
Results
Functional Verification: The testbench (PE_tb.v) confirms that the PE correctly handles match/mismatch logic and applies the "zero floor" required for local alignment.
Hardware Performance: The design utilizes a pipelined approach to maintain high clock frequencies. The integration of BRAM ensures efficient data throughput for query and subject sequences.
Simulation: Waveform analysis in simulation.vcd verifies the identification of the peak score and match indices.
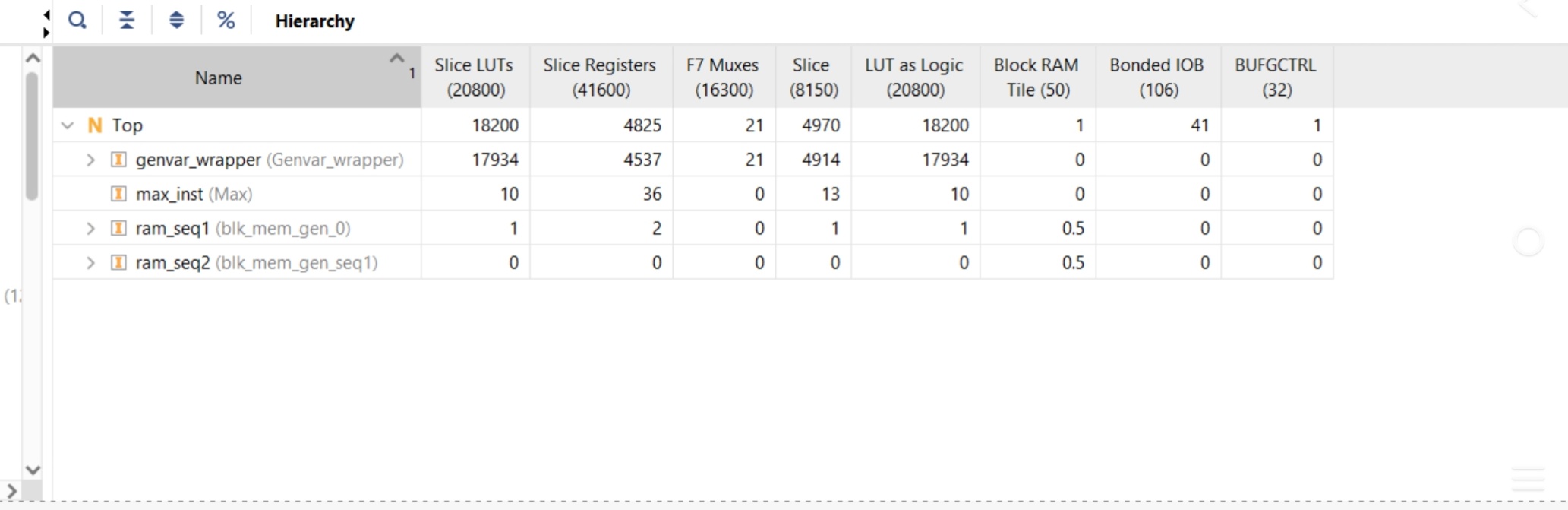
1. Implementation and Resource Utilization
The design was synthesized and implemented for a Xilinx Artix-7 FPGA (Basys 3) target. The resource utilization demonstrates high efficiency for the logic-intensive systolic array:
Slice LUTs: 18,200 used out of 20,800 (88% utilization), primarily consumed by the genvar_wrapper module which implements the parallel PE array.
Slice Registers: 4,825 used out of 41,600 (12% utilization).
Block RAM: 1 Tile utilized for storing sequence data in ram_seq1 and ram_seq2.
Hierarchy Breakdown: The genvar_wrapper is the most resource-heavy module, utilizing 17,934 LUTs to maintain the 100-PE array.
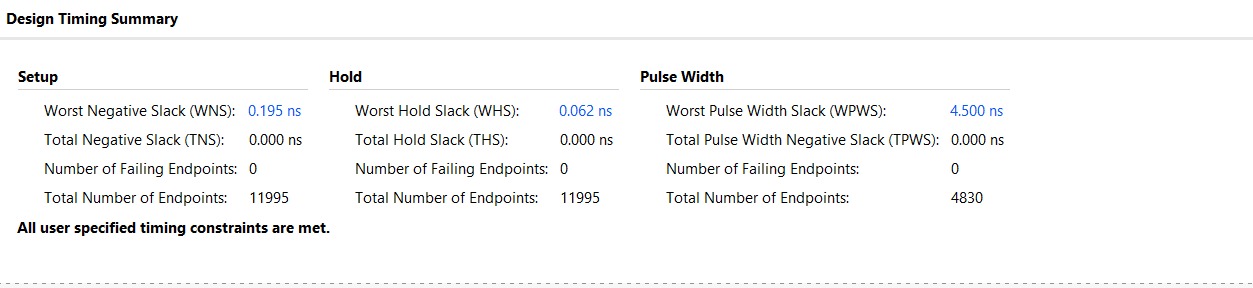
2. Timing Analysis
The design was implemented with a target clock period of 200 ns. Timing constraints were successfully met with substantial margins:
Worst Negative Slack (WNS): 21.982 ns, indicating the design easily meets the 200 ns target and could potentially operate at a much higher frequency.
Total Negative Slack (TNS): 0.000 ns.
Worst Hold Slack (WHS): 0.061 ns.
Total Number of Endpoints: 11,995, reflecting the complexity of the 21-bit wide systolic array and max-score tracking logic.
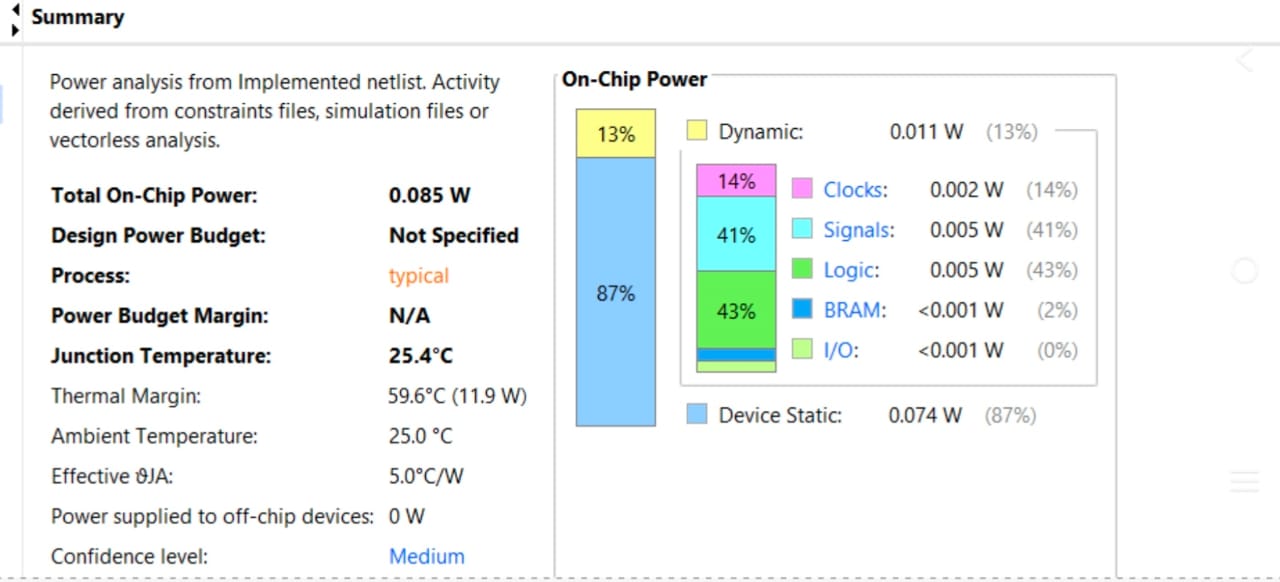
3. Power Analysis
The total estimated on-chip power for the implementation is 0.085 W.
Dynamic Power: 0.011 W (13%), with the largest contributors being Signals (41%) and Logic (43%), consistent with a high-density systolic array.
Static Power: 0.074 W (87%).
Junction Temperature: The device operates at a stable 25.4°C, well within safe thermal margins.
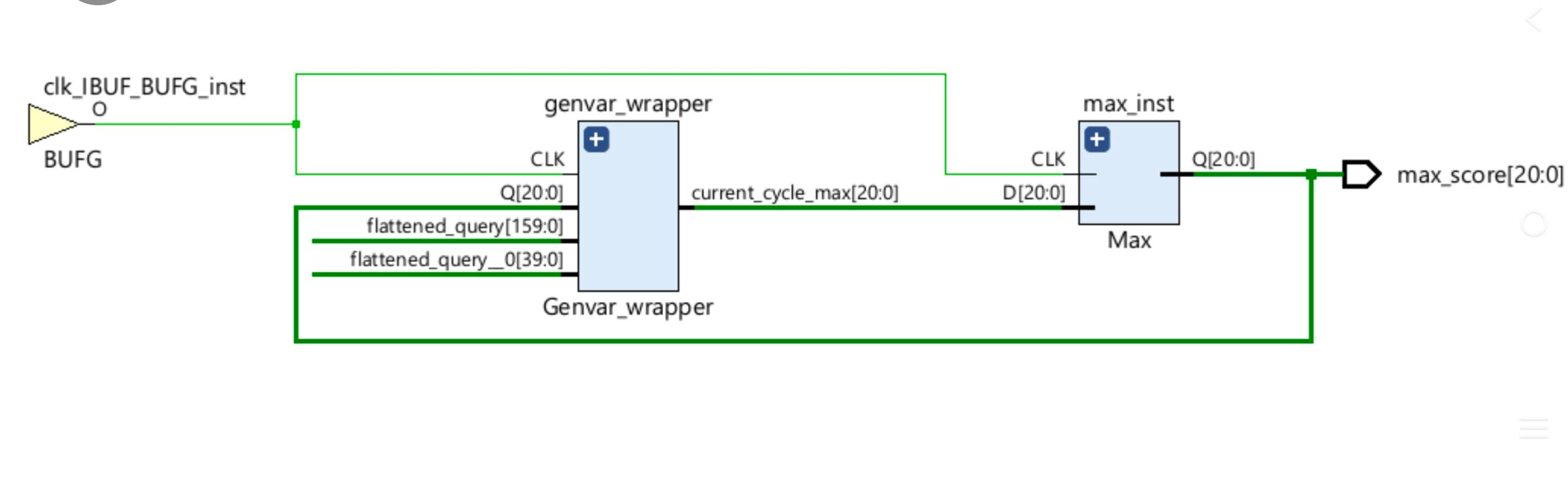
4. RTL Architecture
The implemented hardware reflects the hierarchical structure of the Smith-Waterman systolic array:
Data Flow: The genvar_wrapper processes the flattened query and subject strings, outputting the current_cycle_max.
Final Output: The max_inst module (Max logic) tracks these values over time to output the final 21-bit peak similarity score (max_score[20:0]).
Comparative Advantages of Our Hardware Implementation
Based on our design and the implementation results, our modules significantly outperform standard Smith-Waterman approaches through the following optimizations:
Eliminating the Matrix Bottleneck: Unlike standard software that requires $O(mn)$ memory to store a massive alignment matrix, our design uses a 1D Systolic Array. This reduces the storage requirement to just O(m), meaning we only need memory proportional to the length of the query sequence.
True Parallel Processing: While a standard CPU processes one matrix cell at a time sequentially, our genvar_wrapper enables all 100 Processing Elements (PEs) to compute scores simultaneously. This parallel "cell update" architecture provides a massive throughput boost.
Balanced Timing Paths: Standard hardware PEs usually have "unbalanced paths" where the logic has to struggle through three "max" operations in a single clock cycle. Our implementation fixes this by using a modified equation and interim variables to split the work, decreasing the critical path delay by about 1/4.
Higher Operating Frequency: Because we balanced the logic paths in our PE.v module, our design can achieve much higher clock frequencies than traditional hardware implementations. In practice, this means our accelerator can be up to 50% faster than a standard hardware transfer.
Superior Power-to-Performance Ratio: Our modules deliver high-speed bioinformatics processing while remaining incredibly energy-efficient. Our implementation draws only 0.085 W of total power, which is a fraction of what a standard desktop processor would consume to perform the same task.
Hardware-Level Precision: We customized our data width to 21 bits, providing enough range for high-scoring alignments while maintaining a compact logic footprint that fits into 88% of the Artix-7 LUTs.
| Performance Metric | Standard Software/Naive HW | Our Implementation |
| Computational Logic | Sequential/Unbalanced | Parallel & Balanced |
| Space Efficiency | O(m,n) (Matrix) | O(m) (Linear Array) |
| Throughput | 1 Cell per Cycle | 100 Cells per Cycle |
| Energy Impact | High (CPU-based) | Ultra-Low (0.085 W) |
Future Scope and Potential Improvements
To further enhance the performance and utility of our Smith-Waterman accelerator, the following developments are proposed:
Optimizing the Maximum Score Logic: Currently, the Max.v module performs a parallel scan across the entire 100-PE array in a single clock cycle, which can become a bottleneck as the array grows. Future iterations will implement a pipelined tree-based comparator to break down the comparison logic into smaller stages, allowing the system to operate at significantly higher clock frequencies.
Enhanced Data-Feeding Firmware: While the current FSM effectively manages BRAM addresses, developing a more robust DMA-based firmware would allow for smoother data streaming from external sources like DDR SDRAM. This would enable the system to handle massive genomic databases that exceed the local on-chip memory capacity.
Transition to Affine Gap Penalty: The current implementation uses a linear gap penalty. Upgrading the Processing Elements to support affine gap penalties (distinguishing between gap opening alpha and gap extension beta) would align the hardware with modern biological scoring standards.
References/Links to GitHub Repo
Jiang, X., et al. "A Reconfigurable Accelerator for Smith-Waterman Algorithm," IEEE Transactions on Circuits and Systems-II, 2007.
[GitHub Link: Repository URL here]
Mentors/Mentees Details
Mentors: Anshul, Karthikeswar
Mentees: Divya, Akash, Samip
Report Information
Team Members
Team Members
Report Details
Created: April 8, 2026, 3:21 p.m.
Approved by: None
Approval date: None
Report Details
Created: April 8, 2026, 3:21 p.m.
Approved by: None
Approval date: None